MC2
(Multi-Copy/Monte-Carlo):
A
Macromolecular Docking Program which
Accounts
for Protein Loop Flexibility
![]()
Docking macromolecules with flexible segments.
Bastard K, Thureau A, Lavery R, Prevost C.
J Comput Chem. 2003 Nov 30;24(15):1910-20. (pubmed) (PDF)
We have recently developped a new docking method, termed MC2, which takes into account the loop and side-chain movements at the protein surface during macromolecular association. The program uses a multiple copy representation of the loop coupled with a Monte Carlo process for conformational search.
It is relevant to cases where a flexible loop is suspected to be comprised in the interface, for example by experimental informations or by prior rigid-body docking.
Principle
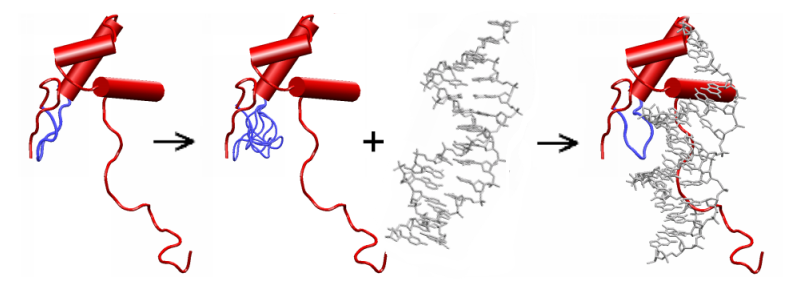
Parts of the protein with fixed main-chain are represented in red. The flexible loop and loop copies are in blue. In the present version, the ligand (in grey) moves as a rigid body.
ResultsApplied on a test-case system, the method was able to predict the structure of the complex at the atomic level and to unambiguously predict the conformation of an interfacial loop.
Among a set of 74 independant runs with various starting geometries, more than 70 % produced a correct loop conformation (< 1.5 Å RMSD for the Calpha), almost 40 % correctly positioned the DNA (< 1.5 Å RMSD on heavy atoms) and 23 % displayed both these characteristics. For the best predictions, the DNA is only at 0.2 Å RMSD from its crystallographic position and the predicted loop conformation differs from that in the reference structure by 0.5 Å for the Calpha and 1 Å for all heavy atoms. Almost 90 % of the correct base/amino acid interactions and 95 % of the native hydrogen bonds are recovered.Since these results were published, the process of loop copy selection has been improved. 24 independent simulations have been carried out with the new parameters and compared with the previous results on the same subset: 96 % (instead of 62 %) produced the correct loop conformation (< 1.5 Å RMSD for the Calpha), 46 % (instead of 50 %) correctly postion the DNA (< 1.5 Å RMSD on heavy atoms) and 42 % (instead of 25 %) associated this two features.
Program availability
When a user friendly version of MC2 will be ready, the software will be distributed under GNU General Public License.
For specific request, please contact us.
![]()
![]()
![]()
![]()
![]()